Gene Expression Note 1
Nur Aziz
1 Profiling Gene Expression from GEO, TCGA, GTEx
Before begin, it’s recommended to read these publications:
- Analysis workflow of publicly available RNA-sequencing datasets
- TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages
- TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data
- New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx
- Unifying cancer and normal RNA sequencing data from different sources
We will make similar results like these :
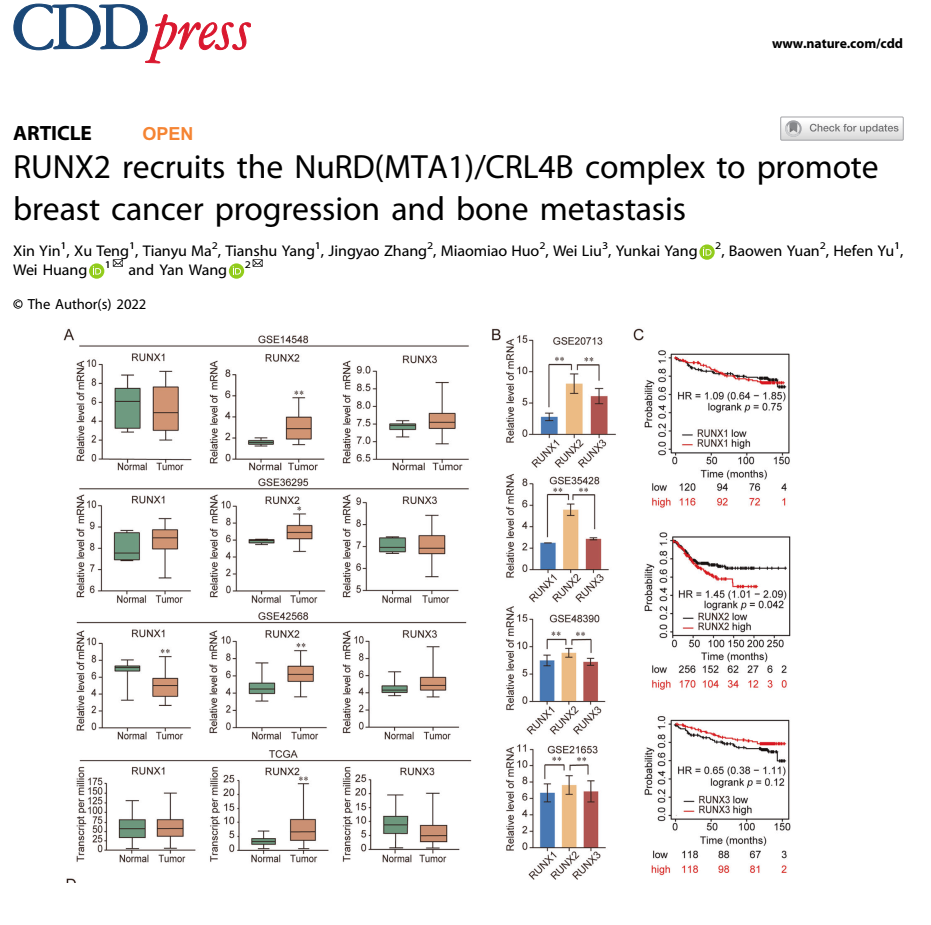
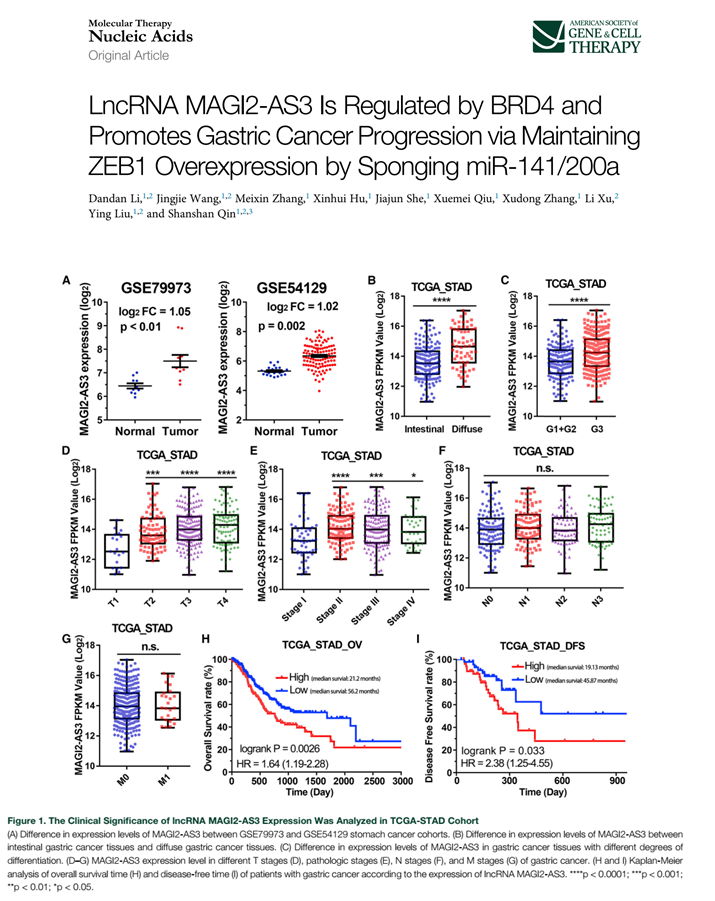
1.1 GEO
1.1.1 Normal vs Tumor
Install and load libraries:
Sys.setenv(LANG='en') #change warning to English
Sys.setenv("VROOM_CONNECTION_SIZE" = 131072 * 10) #prevent connection lost when roaming big chunk
library(GEOquery)
library(limma)
library(tidyverse)
library(dplyr)
library("ggsci")
library(rstatix)
create_dt<-function(x){
DT::datatable(x,
extensions = 'Buttons',
options = list(dom='Blfrtip',
buttons=c('copy', 'csv', 'excel','pdf', 'print'),
lengthMenu=list(c(10,25,50,-1),
c(10,25,50,'All'))))
}To make graph look nice and consistents, I make my own function : * Make color palette for your graph
library(scales)
pallet<-c("#386cb0","#fdb462","#7fc97f","#a6cee3","#fb9a99","#984ea3","#ffff33")
show_col(pallet)
####Aziz' function's theme and color palettes
scale_fill_aziz <- function(...){
library(scales)
discrete_scale("fill","aziz",manual_pal(values = c("#386cb0","#fdb462","#7fc97f","#a6cee3","#fb9a99","#984ea3","#ffff33")), ...)
}
scale_colour_aziz <- function(...){
library(scales)
discrete_scale("colour","aziz",manual_pal(values = c("#386cb0","#fdb462","#7fc97f","#ef3b2c","#662506","#a6cee3","#fb9a99","#984ea3","#ffff33")), ...)
}
theme_Publication <- function(base_size=14, base_family="helvetica") {
library(grid)
library(ggthemes)
(theme_foundation(base_size=base_size, base_family=base_family)
+ theme(plot.title = element_text(face = "bold",
size = rel(1.2), hjust = 0.5),
text = element_text(),
panel.background = element_rect(colour = NA),
plot.background = element_rect(colour = NA),
panel.border = element_rect(colour = NA),
axis.title = element_text(face = "bold",size = rel(1)),
axis.title.y = element_text(angle=90,vjust =2),
axis.title.x = element_text(vjust = -0.2),
axis.text = element_text(),
axis.line = element_line(colour="black"),
axis.ticks = element_line(),
panel.grid.major = element_line(colour="#f0f0f0"),
panel.grid.minor = element_blank(),
legend.key = element_rect(colour = NA),
legend.position = "bottom",
legend.direction = "horizontal",
legend.key.size= unit(0.2, "cm"),
legend.margin = unit(0, "cm"),
legend.title = element_text(face="italic"),
plot.margin=unit(c(10,5,5,5),"mm"),
strip.background=element_rect(colour="#f0f0f0",fill="#f0f0f0"),
strip.text = element_text(face="bold")
))
}For example using ACRG dataset
#### GSE62254 (ACRG)
gset <- getGEO("GSE66229", GSEMatrix =TRUE, AnnotGPL=TRUE)
if (length(gset) > 1) idx <- grep("GPL570", attr(gset, "names")) else idx <- 1
gset <- gset[[idx]]
# make proper column names to match toptable
fvarLabels(gset) <- make.names(fvarLabels(gset))
# group membership for all samples
gsms <- paste0("11100000000000000111110000000000000000100111111111",
"11111111111111111111111111111111111111111111111111",
"11111111111111111111111111111111111111111111111111",
"11111111111111111111111010111101111000000111111111",
"11111111111111111111111111111111111111111111111111",
"11111111111111111111111111111111111111111111111111",
"11111111111111111111111111111111111100000111110000",
"00000000000000000000000000000000000000000000000000")
sml <- strsplit(gsms, split="")[[1]]
# log2 transformation
ex <- exprs(gset)
qx <- as.numeric(quantile(ex, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC <- (qx[5] > 100) ||
(qx[6]-qx[1] > 50 && qx[2] > 0)
if (LogC) { ex[which(ex <= 0)] <- NaN
exprs(gset) <- log2(ex) }
# assign samples to groups and set up design matrix
gs <- factor(sml)
groups <- make.names(c("N","T"))
levels(gs) <- groups
gset$group <- gs
design <- model.matrix(~group + 0, gset)
colnames(design) <- levels(gs)
fit <- lmFit(gset, design) # fit linear model
# set up contrasts of interest and recalculate model coefficients
cts <- paste(groups[1], groups[2], sep="-")
cont.matrix <- makeContrasts(contrasts=cts, levels=design)
fit2 <- contrasts.fit(fit, cont.matrix)
# compute statistics and table of top significant genes
fit2 <- eBayes(fit2, 0.01)
tT <- topTable(fit2, adjust="fdr", sort.by="B", number=250)
tT <- subset(tT, select=c("ID","adj.P.Val","P.Value","t","B","logFC","Gene.symbol","Gene.title"))
# Visualize and quality control test results.
# Build histogram of P-values for all genes. Normal test
# assumption is that most genes are not differentially expressed.
tT2 <- topTable(fit2, adjust="fdr", sort.by="B", number=Inf)
hist(tT2$adj.P.Val, col = "grey", border = "white", xlab = "P-adj",
ylab = "Number of genes", main = "P-adj value distribution")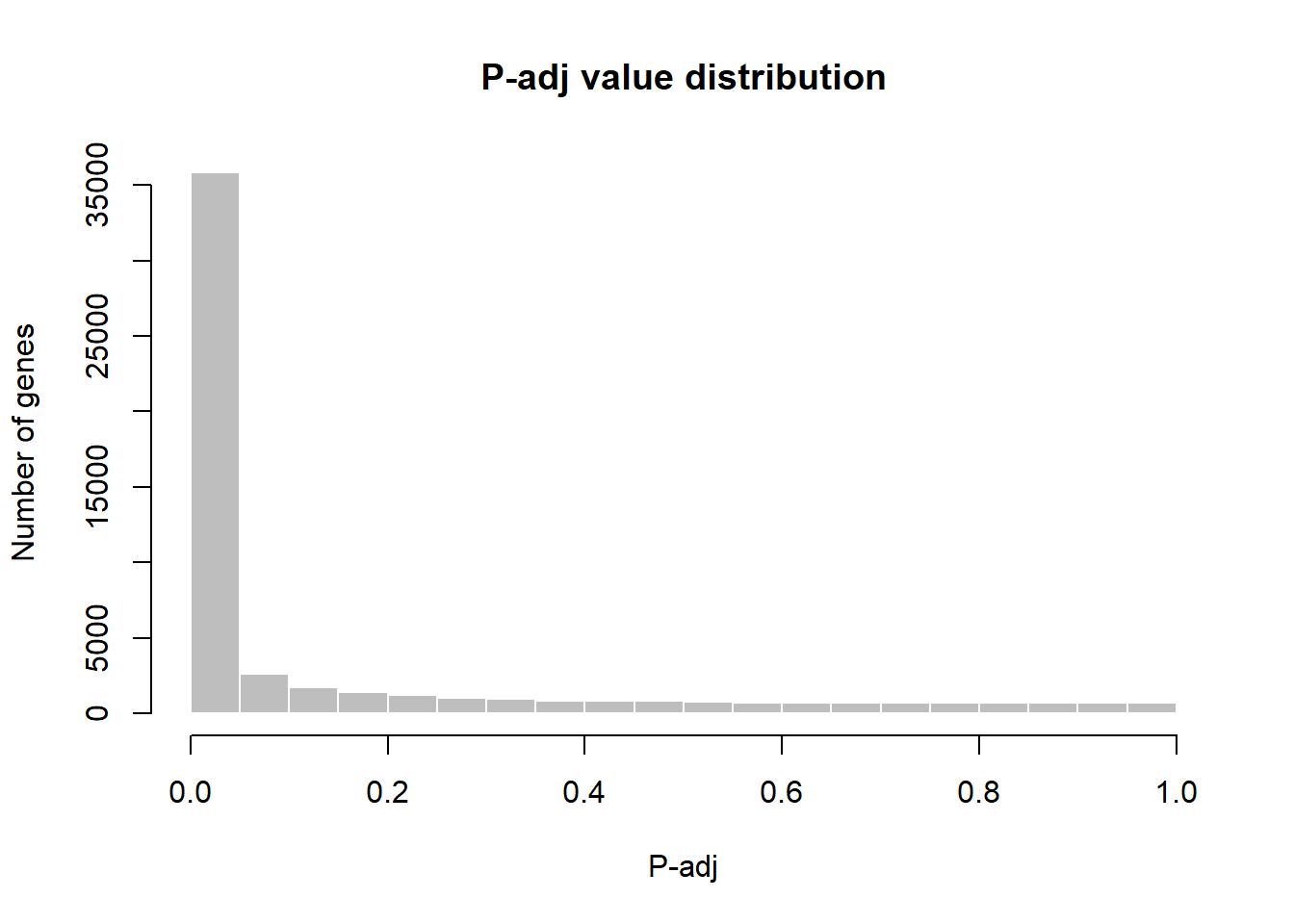
#prepare for only genes passing the threshold
newtT2<-tT2 %>%
filter(adj.P.Val<0.05)
newtT3 <- subset(newtT2,select=c("ID","adj.P.Val","t","B","logFC","Gene.symbol","Gene.title")) #useful for DEGs analysis, but we don't need right now #skip this
#retrieve the expression of your gene in all patient
threegenes<-ex[row.names(ex) %in% c("219617_at", #camkmt
"222013_x_at", #FAM86A
"206445_s_at"),] #PRMT1
head(threegenes)[,1:5]## GSM1523727 GSM1523728 GSM1523729 GSM1523730 GSM1523731
## 206445_s_at 2.957375 3.128566 3.052887 2.916911 2.789613
## 219617_at 1.999631 1.689934 1.806846 1.659223 1.710085
## 222013_x_at 1.758549 1.830104 1.791574 1.750806 1.744396head(t(threegenes)) #transpose the table## 206445_s_at 219617_at 222013_x_at
## GSM1523727 2.957375 1.999631 1.758549
## GSM1523728 3.128566 1.689934 1.830104
## GSM1523729 3.052887 1.806846 1.791574
## GSM1523730 2.916911 1.659223 1.750806
## GSM1523731 2.789613 1.710085 1.744396
## GSM1523732 3.088736 1.715045 1.766945threegenes<-t(threegenes)
GOI<-as.data.frame(threegenes) #GOI = gene of interest
GOI<-GOI %>%
mutate(Patient.ID=row.names(threegenes))
row.names(GOI)<-NULL
GOI<-GOI %>%
select(4,1,2,3)
head(GOI)## Patient.ID 206445_s_at 219617_at 222013_x_at
## 1 GSM1523727 2.957375 1.999631 1.758549
## 2 GSM1523728 3.128566 1.689934 1.830104
## 3 GSM1523729 3.052887 1.806846 1.791574
## 4 GSM1523730 2.916911 1.659223 1.750806
## 5 GSM1523731 2.789613 1.710085 1.744396
## 6 GSM1523732 3.088736 1.715045 1.766945colnames(GOI)[2]<-"PRMT1"
colnames(GOI)[3]<-"CAMKMT"
colnames(GOI)[4]<-"FAM86A"
# download phenotypes
my.gse <- "GSE66229"
if(!file.exists("geo_downloads")) dir.create("geo_downloads")
if(!file.exists("results")) dir.create("results", recursive=TRUE)
my.geo.gse <- getGEO(GEO=my.gse, filename=NULL, destdir="./geo_downloads", GSElimits=NULL, GSEMatrix=TRUE, AnnotGPL=FALSE, getGPL=FALSE)
my.geo.gse <- my.geo.gse[[1]]
my.pdata <- as.data.frame(pData(my.geo.gse), stringsAsFactors=F)
head(my.pdata)## title geo_accession status submission_date
## GSM1523727 T107 GSM1523727 Public on Apr 17 2015 Oct 10 2014
## GSM1523728 T108 GSM1523728 Public on Apr 17 2015 Oct 10 2014
## GSM1523729 T109 GSM1523729 Public on Apr 17 2015 Oct 10 2014
## GSM1523730 N132 GSM1523730 Public on Feb 15 2019 Oct 10 2014
## GSM1523731 N133 GSM1523731 Public on Feb 15 2019 Oct 10 2014
## GSM1523732 N134 GSM1523732 Public on Feb 15 2019 Oct 10 2014
## last_update_date type channel_count source_name_ch1
## GSM1523727 Apr 17 2015 RNA 1 Tumor from patient 107
## GSM1523728 Apr 17 2015 RNA 1 Tumor from patient 108
## GSM1523729 Apr 17 2015 RNA 1 Tumor from patient 109
## GSM1523730 Feb 15 2019 RNA 1 Normal from patient 132
## GSM1523731 Feb 15 2019 RNA 1 Normal from patient 133
## GSM1523732 Feb 15 2019 RNA 1 Normal from patient 134
## organism_ch1 characteristics_ch1 characteristics_ch1.1
## GSM1523727 Homo sapiens tissue: Gastric tumor patient: 107
## GSM1523728 Homo sapiens tissue: Gastric tumor patient: 108
## GSM1523729 Homo sapiens tissue: Gastric tumor patient: 109
## GSM1523730 Homo sapiens tissue: normal gastric tissue patient: 132
## GSM1523731 Homo sapiens tissue: normal gastric tissue patient: 133
## GSM1523732 Homo sapiens tissue: normal gastric tissue patient: 134
## molecule_ch1
## GSM1523727 total RNA
## GSM1523728 total RNA
## GSM1523729 total RNA
## GSM1523730 total RNA
## GSM1523731 total RNA
## GSM1523732 total RNA
## extract_protocol_ch1
## GSM1523727 Samples are processed in parallel in 96-well plates to minimize potential variation. Reaction purification is achieved using magnetic binding beads for cDNA and Qiagen RNeasy kits for cRNA purification.
## GSM1523728 Samples are processed in parallel in 96-well plates to minimize potential variation. Reaction purification is achieved using magnetic binding beads for cDNA and Qiagen RNeasy kits for cRNA purification.
## GSM1523729 Samples are processed in parallel in 96-well plates to minimize potential variation. Reaction purification is achieved using magnetic binding beads for cDNA and Qiagen RNeasy kits for cRNA purification.
## GSM1523730 Samples are processed in parallel in 96-well plates to minimize potential variation. Reaction purification is achieved using magnetic binding beads for cDNA and Qiagen RNeasy kits for cRNA purification.
## GSM1523731 Samples are processed in parallel in 96-well plates to minimize potential variation. Reaction purification is achieved using magnetic binding beads for cDNA and Qiagen RNeasy kits for cRNA purification.
## GSM1523732 Samples are processed in parallel in 96-well plates to minimize potential variation. Reaction purification is achieved using magnetic binding beads for cDNA and Qiagen RNeasy kits for cRNA purification.
## label_ch1 label_protocol_ch1 taxid_ch1
## GSM1523727 biotin standard Affymetrix protocol 9606
## GSM1523728 biotin standard Affymetrix protocol 9606
## GSM1523729 biotin standard Affymetrix protocol 9606
## GSM1523730 biotin standard Affymetrix protocol 9606
## GSM1523731 biotin standard Affymetrix protocol 9606
## GSM1523732 biotin standard Affymetrix protocol 9606
## hyb_protocol
## GSM1523727 GeneChip microarrays are loaded with the fragmented target sample/hybridization buffer mix using standard manual techniques. Arrays are hybridized for 18 hours at 45oC with vigorous mixing. Unbound sample is removed and staining is accomplished through the binding of streptavidin conjugated phycoerythrin to the hybridized target. Excess label is removed. Washing and staining steps are performed by the Affymetrix FS450 fluidics station using standard protocols.
## GSM1523728 GeneChip microarrays are loaded with the fragmented target sample/hybridization buffer mix using standard manual techniques. Arrays are hybridized for 18 hours at 45oC with vigorous mixing. Unbound sample is removed and staining is accomplished through the binding of streptavidin conjugated phycoerythrin to the hybridized target. Excess label is removed. Washing and staining steps are performed by the Affymetrix FS450 fluidics station using standard protocols.
## GSM1523729 GeneChip microarrays are loaded with the fragmented target sample/hybridization buffer mix using standard manual techniques. Arrays are hybridized for 18 hours at 45oC with vigorous mixing. Unbound sample is removed and staining is accomplished through the binding of streptavidin conjugated phycoerythrin to the hybridized target. Excess label is removed. Washing and staining steps are performed by the Affymetrix FS450 fluidics station using standard protocols.
## GSM1523730 GeneChip microarrays are loaded with the fragmented target sample/hybridization buffer mix using standard manual techniques. Arrays are hybridized for 18 hours at 45oC with vigorous mixing. Unbound sample is removed and staining is accomplished through the binding of streptavidin conjugated phycoerythrin to the hybridized target. Excess label is removed. Washing and staining steps are performed by the Affymetrix FS450 fluidics station using standard protocols.
## GSM1523731 GeneChip microarrays are loaded with the fragmented target sample/hybridization buffer mix using standard manual techniques. Arrays are hybridized for 18 hours at 45oC with vigorous mixing. Unbound sample is removed and staining is accomplished through the binding of streptavidin conjugated phycoerythrin to the hybridized target. Excess label is removed. Washing and staining steps are performed by the Affymetrix FS450 fluidics station using standard protocols.
## GSM1523732 GeneChip microarrays are loaded with the fragmented target sample/hybridization buffer mix using standard manual techniques. Arrays are hybridized for 18 hours at 45oC with vigorous mixing. Unbound sample is removed and staining is accomplished through the binding of streptavidin conjugated phycoerythrin to the hybridized target. Excess label is removed. Washing and staining steps are performed by the Affymetrix FS450 fluidics station using standard protocols.
## scan_protocol
## GSM1523727 Arrays are scanned using a GeneChip Scanner 3000 7G with a 48 array autoloader. The scanner maintains the optimal temperature for the arrays prior to and during scanning.
## GSM1523728 Arrays are scanned using a GeneChip Scanner 3000 7G with a 48 array autoloader. The scanner maintains the optimal temperature for the arrays prior to and during scanning.
## GSM1523729 Arrays are scanned using a GeneChip Scanner 3000 7G with a 48 array autoloader. The scanner maintains the optimal temperature for the arrays prior to and during scanning.
## GSM1523730 Arrays are scanned using a GeneChip Scanner 3000 7G with a 48 array autoloader. The scanner maintains the optimal temperature for the arrays prior to and during scanning.
## GSM1523731 Arrays are scanned using a GeneChip Scanner 3000 7G with a 48 array autoloader. The scanner maintains the optimal temperature for the arrays prior to and during scanning.
## GSM1523732 Arrays are scanned using a GeneChip Scanner 3000 7G with a 48 array autoloader. The scanner maintains the optimal temperature for the arrays prior to and during scanning.
## description
## GSM1523727 ACRG Gastric cohort: microarray profiles from 300 gastric tumors and 100 patient matched normal tissue from gastric cancer patients
## GSM1523728 ACRG Gastric cohort: microarray profiles from 300 gastric tumors and 100 patient matched normal tissue from gastric cancer patients
## GSM1523729 ACRG Gastric cohort: microarray profiles from 300 gastric tumors and 100 patient matched normal tissue from gastric cancer patients
## GSM1523730 ACRG Gastric cohort: microarray profiles from 300 gastric tumors and 100 patient matched normal tissue from gastric cancer patients
## GSM1523731 ACRG Gastric cohort: microarray profiles from 300 gastric tumors and 100 patient matched normal tissue from gastric cancer patients
## GSM1523732 ACRG Gastric cohort: microarray profiles from 300 gastric tumors and 100 patient matched normal tissue from gastric cancer patients
## description.1
## GSM1523727 Nugen amplification protocol
## GSM1523728 Nugen amplification protocol
## GSM1523729 Nugen amplification protocol
## GSM1523730 Nugen amplification protocol
## GSM1523731 Nugen amplification protocol
## GSM1523732 Nugen amplification protocol
## data_processing
## GSM1523727 The data were analyzed by RMA with Affymetrix Power Tools package using Affymetrix default analysis settings.
## GSM1523728 The data were analyzed by RMA with Affymetrix Power Tools package using Affymetrix default analysis settings.
## GSM1523729 The data were analyzed by RMA with Affymetrix Power Tools package using Affymetrix default analysis settings.
## GSM1523730 The data were analyzed by RMA with Affymetrix Power Tools package using Affymetrix default analysis settings.
## GSM1523731 The data were analyzed by RMA with Affymetrix Power Tools package using Affymetrix default analysis settings.
## GSM1523732 The data were analyzed by RMA with Affymetrix Power Tools package using Affymetrix default analysis settings.
## platform_id contact_name contact_department
## GSM1523727 GPL570 Michael,,Nebozhyn GpGx/Computational System biology
## GSM1523728 GPL570 Michael,,Nebozhyn GpGx/Computational System biology
## GSM1523729 GPL570 Michael,,Nebozhyn GpGx/Computational System biology
## GSM1523730 GPL570 Michael,,Nebozhyn GpGx/Computational System biology
## GSM1523731 GPL570 Michael,,Nebozhyn GpGx/Computational System biology
## GSM1523732 GPL570 Michael,,Nebozhyn GpGx/Computational System biology
## contact_institute
## GSM1523727 Merck, Inc
## GSM1523728 Merck, Inc
## GSM1523729 Merck, Inc
## GSM1523730 Merck, Inc
## GSM1523731 Merck, Inc
## GSM1523732 Merck, Inc
## contact_address
## GSM1523727 Mail Stop WP53B-120, 770 Sumneytown Pike, Building 53, P.O. Box 4
## GSM1523728 Mail Stop WP53B-120, 770 Sumneytown Pike, Building 53, P.O. Box 4
## GSM1523729 Mail Stop WP53B-120, 770 Sumneytown Pike, Building 53, P.O. Box 4
## GSM1523730 Mail Stop WP53B-120, 770 Sumneytown Pike, Building 53, P.O. Box 4
## GSM1523731 Mail Stop WP53B-120, 770 Sumneytown Pike, Building 53, P.O. Box 4
## GSM1523732 Mail Stop WP53B-120, 770 Sumneytown Pike, Building 53, P.O. Box 4
## contact_city contact_state contact_zip/postal_code contact_country
## GSM1523727 West Point PA 19486 USA
## GSM1523728 West Point PA 19486 USA
## GSM1523729 West Point PA 19486 USA
## GSM1523730 West Point PA 19486 USA
## GSM1523731 West Point PA 19486 USA
## GSM1523732 West Point PA 19486 USA
## supplementary_file
## GSM1523727 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM1523nnn/GSM1523727/suppl/GSM1523727_INT201201-10-1_107T_HG-U133_Plus_2_.CEL.gz
## GSM1523728 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM1523nnn/GSM1523728/suppl/GSM1523728_INT201201-10-1_108T_HG-U133_Plus_2_.CEL.gz
## GSM1523729 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM1523nnn/GSM1523729/suppl/GSM1523729_INT201201-10-1_109T_HG-U133_Plus_2_.CEL.gz
## GSM1523730 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM1523nnn/GSM1523730/suppl/GSM1523730_INT201201-10-1_132N_HG-U133_Plus_2_.CEL.gz
## GSM1523731 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM1523nnn/GSM1523731/suppl/GSM1523731_INT201201-10-1_133N_HG-U133_Plus_2_.CEL.gz
## GSM1523732 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM1523nnn/GSM1523732/suppl/GSM1523732_INT201201-10-1_134N_HG-U133_Plus_2_.CEL.gz
## data_row_count patient:ch1 tissue:ch1
## GSM1523727 54675 107 Gastric tumor
## GSM1523728 54675 108 Gastric tumor
## GSM1523729 54675 109 Gastric tumor
## GSM1523730 54675 132 normal gastric tissue
## GSM1523731 54675 133 normal gastric tissue
## GSM1523732 54675 134 normal gastric tissuephenotype<-my.pdata %>%
select(2,1)
row.names(phenotype)<-NULL
#extract Normal or Tumor tissues
phenotype<-phenotype %>%
mutate(Tissue.type=substr(title,1,1)) %>%
select(1,3)
colnames(phenotype)[1]<-"Patient.ID"
# merge expression and phenotype dataframe
acrg.merged<-phenotype %>%
left_join(GOI)
create_dt(acrg.merged)1.1.2 Plotting
acrg<-acrg.merged %>%
mutate(Tissue2 = case_when(Tissue.type == 'N' ~ 'Normal',
Tissue.type == 'T' ~ 'Tumor'))
acrg$Tissue2 <- factor(acrg$Tissue2, levels = c("Normal", "Tumor"))
GSEacrg1<-ggplot(acrg,aes(x=Tissue2,y=FAM86A,fill=Tissue2))+
labs(y=expression('FAM86A expression (log'[10]*")"),x=NULL)+
stat_boxplot(geom ='errorbar',width=.2) +
geom_jitter(colour='gray90',alpha=.8,width = .2)+
geom_boxplot(width=.6)+
theme_Publication()+
scale_fill_aziz()+
scale_y_continuous(expand=expand_scale(mult = c(0.1,0.2)))+
theme(axis.text.x = element_text(),
legend.position = 'none',
axis.title.x = element_blank(),
panel.grid.major.x = element_blank(),
aspect.ratio = 1.85)
GSEacrg2<-ggplot(acrg,aes(x=Tissue2,y=CAMKMT,fill=Tissue2))+
labs(y=expression('CAMKMT expression (log'[10]*")"),x=NULL)+
stat_boxplot(geom ='errorbar',width=.2) +
geom_jitter(colour='gray90',alpha=.8,width = .2)+
geom_boxplot(width=.6)+
theme_Publication()+
scale_fill_aziz()+
scale_y_continuous(expand=expand_scale(mult = c(0.1,0.2)))+
theme(axis.text.x = element_text(),
legend.position = 'none',
axis.title.x = element_blank(),
panel.grid.major.x = element_blank(),
aspect.ratio = 1.85)
GSEacrg3<-ggplot(acrg,aes(x=Tissue2,y=PRMT1,fill=Tissue2))+
labs(y=expression('PRMT1 expression (log'[10]*")"),x=NULL)+
stat_boxplot(geom ='errorbar',width=.2) +
geom_jitter(colour='gray90',alpha=.8,width = .2)+
geom_boxplot(width=.6)+
theme_Publication()+
scale_fill_aziz()+
scale_y_continuous(expand=expand_scale(mult = c(0.1,0.2)))+
theme(axis.text.x = element_text(),
legend.position = 'none',
axis.title.x = element_blank(),
panel.grid.major.x = element_blank(),
aspect.ratio = 1.85)
library(gridExtra)
grid.arrange(GSEacrg1,GSEacrg2,GSEacrg3,ncol=3) 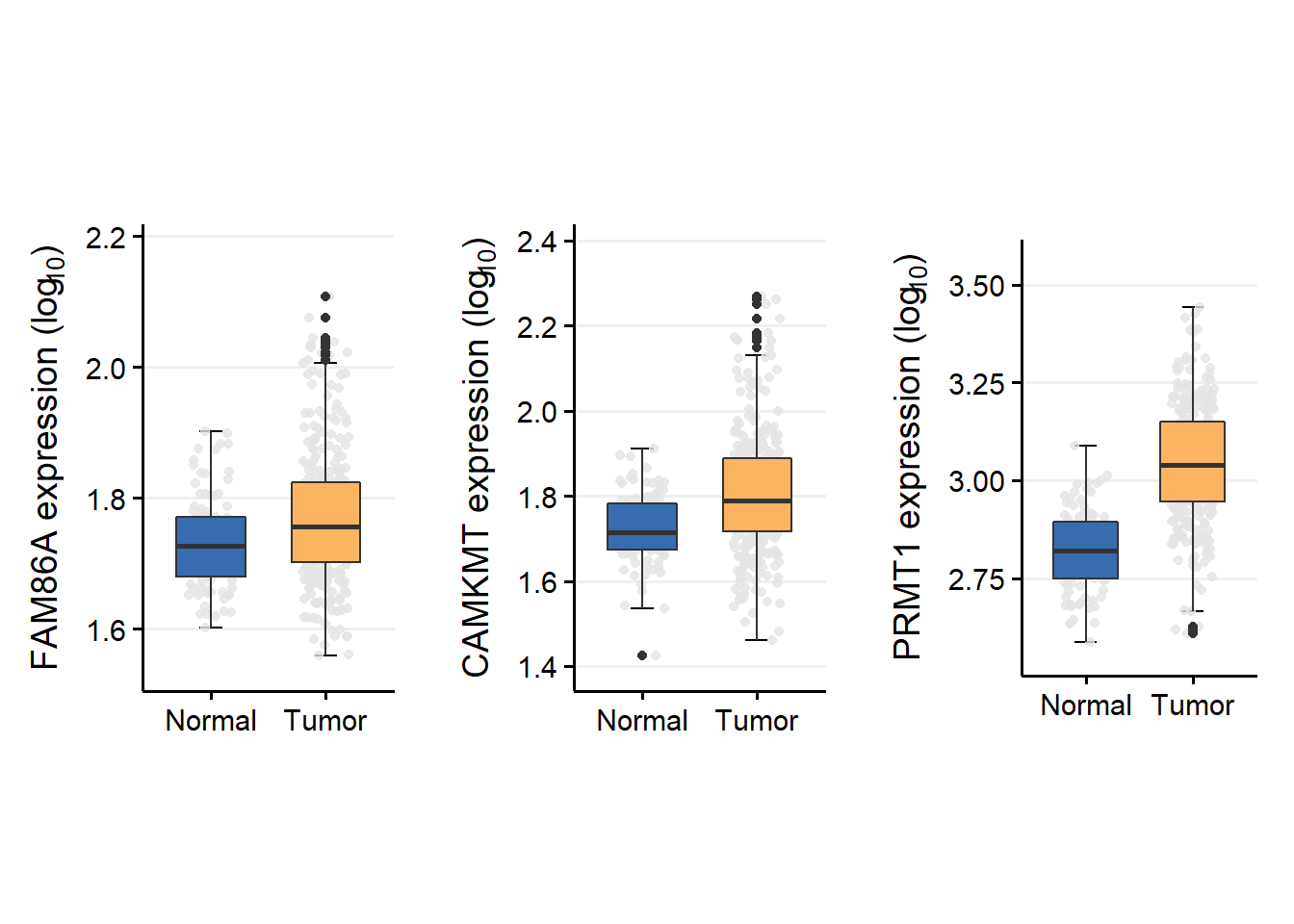
1.1.3 Statistical analysis
wilcox.test(FAM86A ~ Tissue2, data = acrg,
exact = FALSE)##
## Wilcoxon rank sum test with continuity correction
##
## data: FAM86A by Tissue2
## W = 11848, p-value = 0.001646
## alternative hypothesis: true location shift is not equal to 01.2 Unify GTEx NAT and Tumor TCGA
- datasets retrieve from [Nature scientific data] (https://doi.org/10.1038/sdata.2018.61)
- first load the all 3 datasets (tcga normal, tcga tumor, and gtex -> FPKM normalized Combat batch correction)
stomach.rsem.fpkm.gtex <- read.delim("docs/Gene_Exp_1/stomach-rsem-fpkm-gtex.txt")
stad.rsem.fpkm.tcga.t <- read.delim("docs/Gene_Exp_1/stad-rsem-fpkm-tcga-t.txt")
stad.rsem.fpkm.tcga <- read.delim("docs/Gene_Exp_1/stad-rsem-fpkm-tcga.txt")
df.gtex<-stomach.rsem.fpkm.gtex
df.t.tcga<-stad.rsem.fpkm.tcga.t
df.n.tcga<-stad.rsem.fpkm.tcga
library(tidyverse)
library(dplyr)
Sys.setenv(LANG='en')
############GTEx###############
colnames(df.gtex)[1:5]## [1] "Hugo_Symbol" "Entrez_Gene_Id"
## [3] "GTEX.QCQG.0526.SM.48U2A" "GTEX.QDVJ.1426.SM.48U1Y"
## [5] "GTEX.13111.1226.SM.5GCNC"df.gtex.FAM86A<-df.gtex %>%
filter(Hugo_Symbol == 'FAM86A')
head(df.gtex.FAM86A)[2]## Entrez_Gene_Id
## 1 196483dim(df.gtex.FAM86A)## [1] 1 194#transposing
df.gtex.FAM86A<-df.gtex.FAM86A %>%
gather(Patients,FPKM,3:ncol(.))
dim(df.gtex.FAM86A)## [1] 192 4head(df.gtex.FAM86A)[1:4]## Hugo_Symbol Entrez_Gene_Id Patients FPKM
## 1 FAM86A 196483 GTEX.QCQG.0526.SM.48U2A 224.97
## 2 FAM86A 196483 GTEX.QDVJ.1426.SM.48U1Y 220.32
## 3 FAM86A 196483 GTEX.13111.1226.SM.5GCNC 121.79
## 4 FAM86A 196483 GTEX.S4Z8.1226.SM.4AD6W 185.11
## 5 FAM86A 196483 GTEX.ZDYS.1926.SM.5HL59 171.45
## 6 FAM86A 196483 GTEX.144GM.1726.SM.5O9AS 185.11df.gtex.FAM86A.log2<-df.gtex.FAM86A %>%
mutate(log2FPKM=log(FPKM,base = 2))
head(df.gtex.FAM86A.log2)[1:5]## Hugo_Symbol Entrez_Gene_Id Patients FPKM log2FPKM
## 1 FAM86A 196483 GTEX.QCQG.0526.SM.48U2A 224.97 7.813589
## 2 FAM86A 196483 GTEX.QDVJ.1426.SM.48U1Y 220.32 7.783457
## 3 FAM86A 196483 GTEX.13111.1226.SM.5GCNC 121.79 6.928252
## 4 FAM86A 196483 GTEX.S4Z8.1226.SM.4AD6W 185.11 7.532239
## 5 FAM86A 196483 GTEX.ZDYS.1926.SM.5HL59 171.45 7.421644
## 6 FAM86A 196483 GTEX.144GM.1726.SM.5O9AS 185.11 7.532239#######NAT###########
colnames(df.n.tcga)[1:5]## [1] "Hugo_Symbol" "Entrez_Gene_Id"
## [3] "TCGA.BR.6564.11A.01R.1884.13" "TCGA.HU.A4GH.11A.11R.A36D.31"
## [5] "TCGA.IN.8663.11A.01R.2402.13"df.n.tcga.FAM86A<-df.n.tcga %>%
filter(Hugo_Symbol == 'FAM86A')
head(df.n.tcga.FAM86A)[2]## Entrez_Gene_Id
## 1 196483dim(df.n.tcga.FAM86A)## [1] 1 35#transposing
df.n.tcga.FAM86A<-df.n.tcga.FAM86A %>%
gather(Patients,FPKM,3:ncol(.))
dim(df.n.tcga.FAM86A)## [1] 33 4head(df.n.tcga.FAM86A)[1:4]## Hugo_Symbol Entrez_Gene_Id Patients FPKM
## 1 FAM86A 196483 TCGA.BR.6564.11A.01R.1884.13 220.32
## 2 FAM86A 196483 TCGA.HU.A4GH.11A.11R.A36D.31 202.66
## 3 FAM86A 196483 TCGA.IN.8663.11A.01R.2402.13 166.73
## 4 FAM86A 196483 TCGA.HU.8238.11A.01R.2343.13 163.28
## 5 FAM86A 196483 TCGA.BR.7716.11A.01R.2055.13 249.73
## 6 FAM86A 196483 TCGA.FP.7735.11A.01R.2055.13 256.78df.n.tcga.FAM86A.log2<-df.n.tcga.FAM86A %>%
mutate(log2FPKM=log(FPKM,base = 2))
head(df.n.tcga.FAM86A.log2)[1:5]## Hugo_Symbol Entrez_Gene_Id Patients FPKM log2FPKM
## 1 FAM86A 196483 TCGA.BR.6564.11A.01R.1884.13 220.32 7.783457
## 2 FAM86A 196483 TCGA.HU.A4GH.11A.11R.A36D.31 202.66 7.662918
## 3 FAM86A 196483 TCGA.IN.8663.11A.01R.2402.13 166.73 7.381370
## 4 FAM86A 196483 TCGA.HU.8238.11A.01R.2343.13 163.28 7.351204
## 5 FAM86A 196483 TCGA.BR.7716.11A.01R.2055.13 249.73 7.964225
## 6 FAM86A 196483 TCGA.FP.7735.11A.01R.2055.13 256.78 8.004389#######Tumor###########
colnames(df.t.tcga)[1:5]## [1] "Hugo_Symbol" "Entrez_Gene_Id"
## [3] "TCGA.BR.6457.01A.21R.1802.13" "TCGA.VQ.A8PD.01A.11R.A414.31"
## [5] "TCGA.BR.6803.01A.11R.1884.13"df.t.tcga.FAM86A<-df.t.tcga %>%
filter(Hugo_Symbol == 'FAM86A')
head(df.t.tcga.FAM86A)[2]## Entrez_Gene_Id
## 1 196483dim(df.t.tcga.FAM86A)## [1] 1 382#transposing
df.t.tcga.FAM86A<-df.t.tcga.FAM86A %>%
gather(Patients,FPKM,3:ncol(.))
dim(df.t.tcga.FAM86A)## [1] 380 4head(df.t.tcga.FAM86A)[1:4]## Hugo_Symbol Entrez_Gene_Id Patients FPKM
## 1 FAM86A 196483 TCGA.BR.6457.01A.21R.1802.13 325.29
## 2 FAM86A 196483 TCGA.VQ.A8PD.01A.11R.A414.31 215.77
## 3 FAM86A 196483 TCGA.BR.6803.01A.11R.1884.13 346.29
## 4 FAM86A 196483 TCGA.VQ.A8E3.01A.11R.A39E.31 277.20
## 5 FAM86A 196483 TCGA.CG.5718.01A.11R.1602.13 351.14
## 6 FAM86A 196483 TCGA.VQ.A8E2.01A.11R.A36D.31 356.05df.t.tcga.FAM86A.log2<-df.t.tcga.FAM86A %>%
mutate(log2FPKM=log(FPKM,base = 2))
head(df.t.tcga.FAM86A.log2)[1:5]## Hugo_Symbol Entrez_Gene_Id Patients FPKM log2FPKM
## 1 FAM86A 196483 TCGA.BR.6457.01A.21R.1802.13 325.29 8.345583
## 2 FAM86A 196483 TCGA.VQ.A8PD.01A.11R.A414.31 215.77 7.753350
## 3 FAM86A 196483 TCGA.BR.6803.01A.11R.1884.13 346.29 8.435837
## 4 FAM86A 196483 TCGA.VQ.A8E3.01A.11R.A39E.31 277.20 8.114783
## 5 FAM86A 196483 TCGA.CG.5718.01A.11R.1602.13 351.14 8.455903
## 6 FAM86A 196483 TCGA.VQ.A8E2.01A.11R.A36D.31 356.05 8.475936###############combine the 3 data to 1###########
s.gtex<-df.gtex.FAM86A.log2 %>%
mutate(data='GTEx') %>%
dplyr::select('data','log2FPKM')
head(s.gtex)[1:2]## data log2FPKM
## 1 GTEx 7.813589
## 2 GTEx 7.783457
## 3 GTEx 6.928252
## 4 GTEx 7.532239
## 5 GTEx 7.421644
## 6 GTEx 7.532239dim(s.gtex)## [1] 192 2s.nor<-df.n.tcga.FAM86A.log2 %>%
mutate(data='NAT') %>%
dplyr::select('data','log2FPKM')
s.tumor<-df.t.tcga.FAM86A.log2 %>%
mutate(data='Tumor') %>%
dplyr::select('data','log2FPKM')
#binding rows
unify.FAM86A<-rbind(s.gtex,s.nor,s.tumor)
head(unify.FAM86A)[1:2]## data log2FPKM
## 1 GTEx 7.813589
## 2 GTEx 7.783457
## 3 GTEx 6.928252
## 4 GTEx 7.532239
## 5 GTEx 7.421644
## 6 GTEx 7.532239dim(unify.FAM86A)## [1] 605 2count(unify.FAM86A,data)## data n
## 1 GTEx 192
## 2 NAT 33
## 3 Tumor 3801.2.1 Plotting
a<-unify.FAM86A %>%
mutate('source'= case_when(data == 'GTEx' ~ 'GTEx',
data == 'NAT' ~ 'Normal TCGA',
data == 'Tumor' ~ 'Tumor TCGA'))
a %>%
ggplot(aes(x=source,y=log2FPKM,fill=source))+
stat_boxplot(geom ='errorbar',width=.2) +
geom_jitter(colour='gray90',alpha=.8,width = .2)+
geom_boxplot(width=.6)+
theme_Publication()+
scale_fill_aziz()+
scale_y_continuous(expand=expand_scale(mult = c(0.1,0.2)))+
theme(axis.text.x = element_text(angle=45,hjust=1,vjust=1),
legend.position = 'none',
axis.title.x = element_blank(),
panel.grid.major.x = element_blank(),
aspect.ratio = 1.85)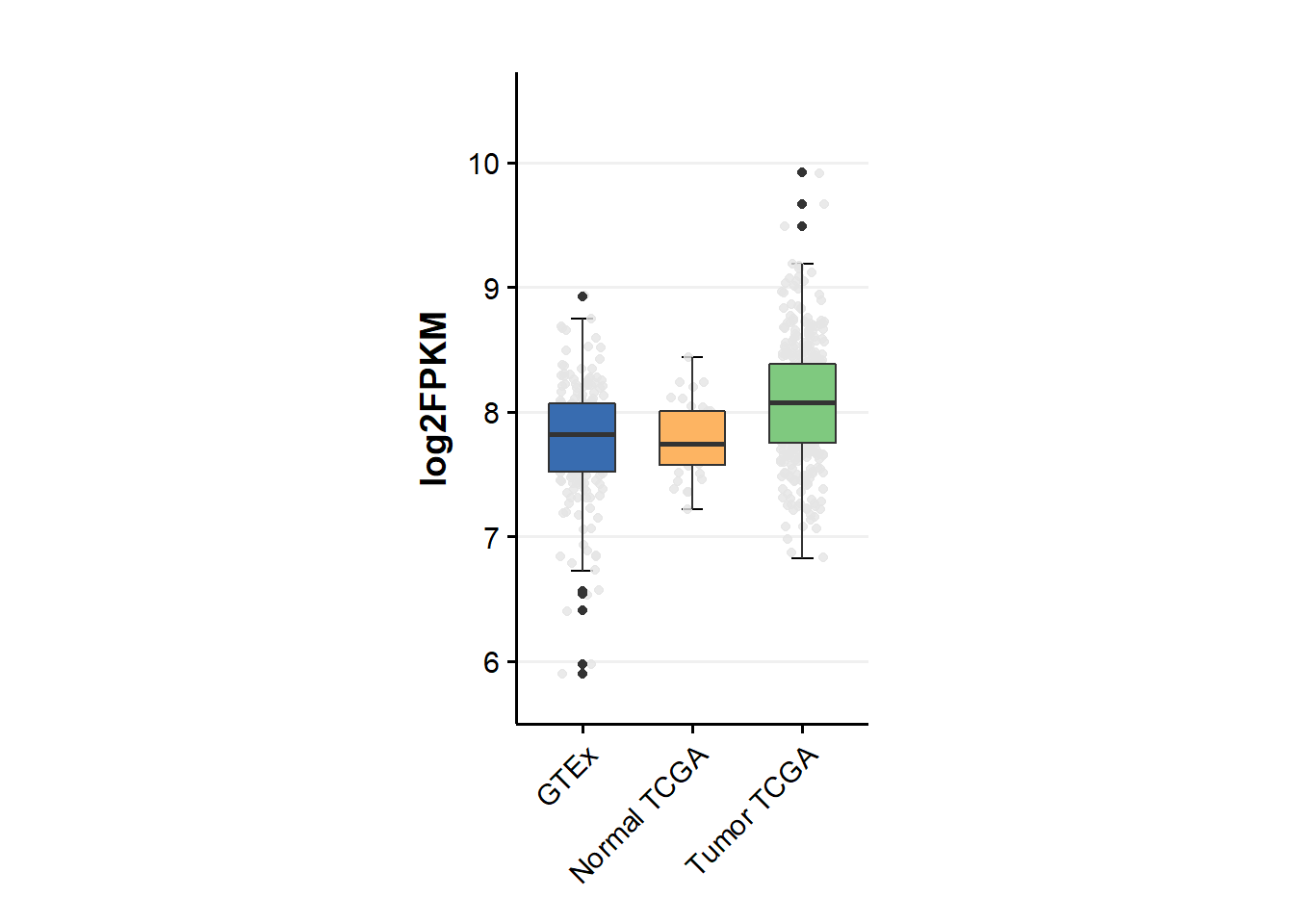
1.2.2 Statistical analysis
kruskal.test(log2FPKM ~ source, data = a)##
## Kruskal-Wallis rank sum test
##
## data: log2FPKM by source
## Kruskal-Wallis chi-squared = 55.539, df = 2, p-value = 8.706e-13pairwise.wilcox.test(a$log2FPKM, a$source,paired=FALSE,
p.adjust.method = "BH")##
## Pairwise comparisons using Wilcoxon rank sum test with continuity correction
##
## data: a$log2FPKM and a$source
##
## GTEx Normal TCGA
## Normal TCGA 0.63 -
## Tumor TCGA 2.1e-11 8.9e-05
##
## P value adjustment method: BH